New research from the Denmark laboratory uses a computer-driven workflow and machine learning to develop a more efficient and versatile method of selective catalyst prediction. The research was conducted by Prof. Scott E. Denmark and graduate students Andrew F. Zahrt, Jeremy J. Henle, Brennan T. Rose, William T. Darrow, and postdoc Dr. Yang Wang.
For centuries, chemists have been preoccupied with the question of how molecular structure relates to function, be it an antibiotic, fragrance, flame retardant, pigment, or vitamin—essentially, every natural and man-made object on Earth.
A corollary to this endeavor is the question of how to manipulate molecular structure to optimize the performance of the object itself.
One of the most spectacular triumphs of modern synthetic chemistry is the ability to make and modify molecules with surgical precision at the atomic level. This accomplishment is enabled by the invention of highly specialized reagents and catalysts that can break and forge new chemical bonds with a stunning level of selectivity in the face of many possibilities.
“Unfortunately,” said Denmark, the Reynold C. Fuson Professor of Chemistry, “the development of these ‘magical’ agents is an extremely challenging enterprise that, even in the 21st century, relies on intuition and empirical reasoning. Through often arduous and time-consuming trial and error, even the most proficient experts can take years to invent a new reagent or catalyst for a specific operation.
 With generous support from the W.M. Keck Foundation, the Denmark laboratory has developed a computationally-guided workflow to accelerate the identification and optimization of catalysts.
With generous support from the W.M. Keck Foundation, the Denmark laboratory has developed a computationally-guided workflow to accelerate the identification and optimization of catalysts.
A critical feature of the new approach is the creation of a virtual library of thousands of hypothetical catalyst structures that encompass an enormous diversity of molecular properties. Generating an extremely diverse library maximizes the probability of creating one or more structures with the ideal properties to effect the desired transformation with optimum performance.
The challenge, however, is finding the needle in that massive haystack.
“To do this, Denmark said, “we identify a subset of library members that represent the chemical diversity to the greatest extent possible, much in the way our 535 members of Congress represent the 350 million citizens of the United States. The ‘representatives’ constitute a ‘training set’ from which we collect experimental data on a given transformation. That training set data is used as input into a machine-learning algorithm, which can can find patterns in large amounts of information that are beyond the cognitive limit of humans.”
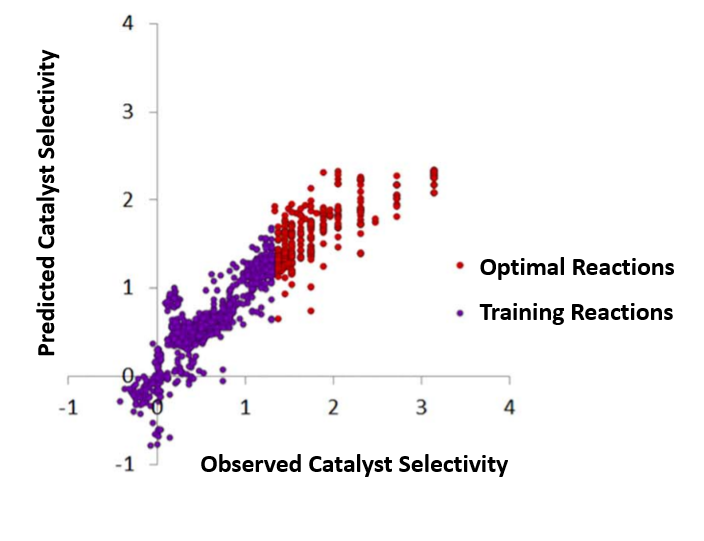
The mathematical equations generated by the algorithms then predict the behavior of each catalyst in the virtual library, making it possible to select the best catalyst for the transformation under study.
In their seminal publication, Denmark’s team demonstrates the power of this approach by using a set of non-optimal data to predict optimal catalysts for a test reaction. The model accurately ranked the catalysts in order of their selectivity, successfully simulating the optimization of a heretofore unoptimized reaction.
After a decade of failures, this accomplishment represents a successful culmination and harbinger of exciting avenues to pursue for decades to come.
The research was published in the January 18, 2019 issue of Science magazine. Read the full paper here.