A research team led by University of Illinois Urbana-Champaign chemists has developed an easy-to-use intuitive tool that could help in the design of new mechanophores with a range of practical applications in materials science and organic synthesis due to their ability to change physically or chemically in response to force.
A mechanophore discovered a few years ago by Illinois chemists, including Prof. Jeffrey Moore and graduate student Yunyan Sun, can release controlled amounts of carbon monoxide when triggered by mechanical force, which can potentially be used in the human body as a drug to treat diseases and conditions. Called NEO, it depends on the breaking of carbon-carbon bonds, which is common in a variety of mechanochemical transformations.
But a major challenge in studying mechanophores is understanding and predicting the reactivity of breaking the C-C bond. Typically, that requires a lot of experimentation and calculation due to the unpredictable vectoral nature of forces like pushing or pulling.
A research team that includes Moore and Sun and researchers at MIT and Duke University has developed a simple and intuitive tool that can predict without lengthy experimentation and calculation the reactivity of C-C bonds when designing mechanophores. Their paper, “The Tension Activated Carbon-Carbon Bond,” just published in the journal Chem, explains their work on this tool, which reports a new computational model and its experimental validation.
According to the researchers, this tool could ultimately lead to the discovery of new mechanically responsive materials and enhance understanding of structure-reactivity relationships, leading to advancements in the field of mechanochemistry.
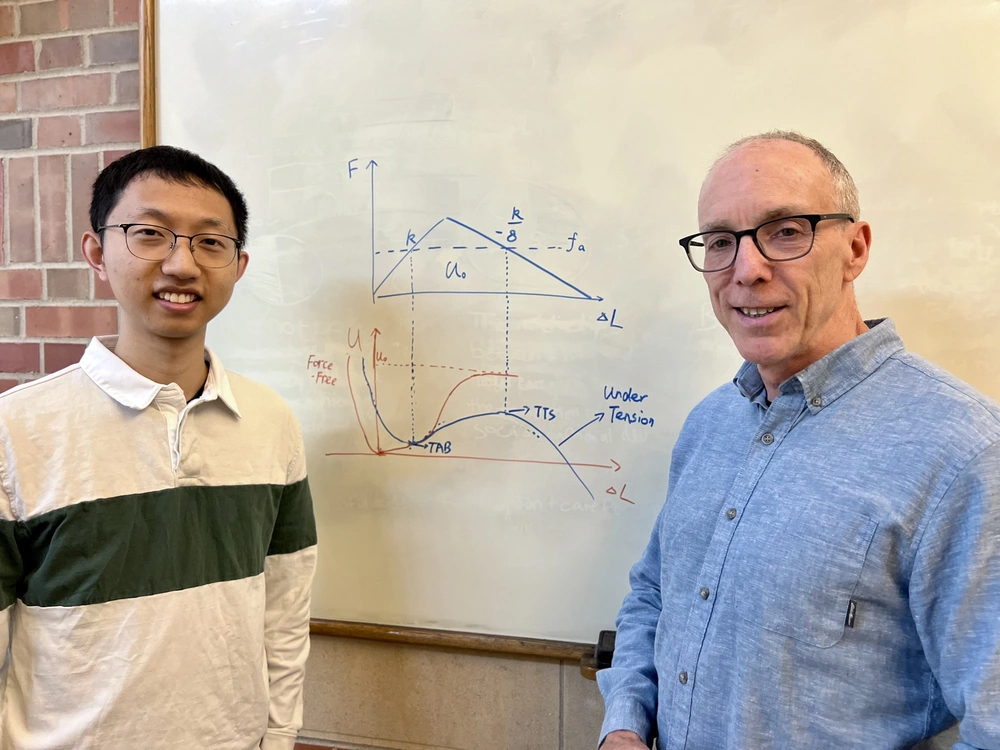
Sun said the tool is derived from the Morse Potential, an interatomic interaction model for the potential energy of a diatomic molecule and a classic chemistry model familiar to freshman level college students.
“It's very, very simple derivation. Everyone can do it,” Sun said of this mnemonic tool, called the tension model of bond activation (TMBA).
By constructing the restoring force triangle, TMBA captures the mechanochemical activation of C–C bonds in complex molecules using two easily computed parameters – effective force constant and reaction energy – as the key molecular features that govern mechanochemical kinetics.
Their triangle helps make sense of complex computations, transforming the results into useful information.
“These calculations are quantitatively accurate but difficult to intuit. Our triangle is a way to find molecular features and insight from the results, and therefore gain a better understanding of what this computational model is telling you,” Moore explained.
Sun said providing an intuitive tool like this is very important.
“There are always high-level complicated computations that can help you to some extent predict and understand things, but in most cases it's not super helpful to start with those. You need something that's intuitive and simple to help you, to help guide you designing mechanophores. Our model really helps you understand what’s going on with the complicated calculations, but you just see the results,” Sun said.

Chemistry is taught and understood using simplified models that boil down complex interactions into simple to understand concepts, said study co-author Ilia Kevlishvili, a postdoctoral researcher in the lab of Heather J. Kulik, professor of chemical engineering and chemistry at MIT. Models have been developed to describe things ranging from molecular structures (Lewis structures) to reaction thermodynamics (HSAB) and kinetics (Hammond’s postulate, Marcus theory, Woodward-Hoffmann rules), Kevlishvili said.
“Similarly, we have developed a simple framework to rationalize and understand the reactivity of mechanically activated bonds that is both easy to understand and quantitatively informative,” Kevlishvili explained. “The model itself can be used to discover new modes of reactivity giving rise to new functional materials. Furthermore, the concept of productive mechanochemistry is relatively fresh and providing an easily understandable model for how mechanical stress can lead to precise reactivity is important.”
The idea for this work began with Sun and Kevlishvili studying a series of NEO mechanophores and noticing an unexpected reactivity trend. Diving deeper they realized there could be key parameters that correlate to a common trend among some mechanophores.
“And then we started to wonder if this is a trend that could be generalized to more general cases of mechanophores, and then we started to wonder if there is something more fundamental behind it that could help the whole field of mechanochemistry,” Sun said.
Moore said the initial idea turned into a great collaboration with researchers at MIT and Duke who are their partners in MONET, the Center for the Chemistry of Molecularly Optimized NETworks, which is funded by the NSF Centers for Chemical Innovation program. Moore said some of the concepts initiated from ideas that came out of Prof. Steve Craig's lab at Duke.
“The concept of the CCI really worked in this case,” Moore said.
Through extensive experimentation and computation, the researchers studied four NEO mechanophores to determine the two key molecular features and then create their linear model, which accurately predicted the transition force required for C–C bond activation in over 30 known mechanophores. The result of their work represents a highly accessible tool that can be used in large-scale screening to predict the mechanochemical reactivity C–C bond in the design of mechanophores.
Kevlishvili said CCIs like MONET provide an immense opportunity to bring together individuals with expertise in different areas.

“This project was my first introduction to the field of mechanochemistry but because I was working closely with Yunyan, Jeff, and Steve, I did not miss a beat and I was quickly caught up. This work was only made possible due to multiple groups coming together, as each piece of data gained from each group was crucial to build the TMBA framework,” Kevlishvili said.
Stephen L. Craig, professor of chemistry at Duke, and Tatiana Kouznetsova, lab research analyst at Duke, provided single-molecule studies of a series of mechanochemical reactions for the project. Craig said he appreciates how Sun and Kevlishvili successfully brought together their own perspectives and complementary technical approaches in this work.
“It was a pleasure to be recruited into this collaboration,” Craig said. “I am excited to see if the method provides an efficient route to the discovery of efficient mechanophores.”
Also, Moore said, their model is distilled down into something that is very teachable with a conceptual framework that could be textbook knowledge in the future.
“Because of that teachability, it brings understanding at a level that could be taught from a freshman level on,” he said. “You always thought the C-C bond was indestructible, but it's not. If you design your molecule just right so that you can bring force to that bond and if you stretch it to a certain point and there's an alternative reaction pathway available, it might take that pathway and you have a new chemical compound or may have new functionality.”
Funding for this work was supported by the NSF Center for the Chemistry of Molecularly Optimized Networks (MONET).
In the news:
https://phys.org/news/2024-06-simple-tool-discovery-mechanically-respon…
Editor’s notes
DOI: https://doi.org/10.1016/j.chempr.2024.05.012
Contact Jeffrey Moore at jsmoore@illinois.edu
Contact Yunyan Sun at yunyans2@illinois.edu
Contact Ilia Kevlishvili at iliak@mit.edu
Contact Stephen Craig at stephen.craig@duke.edu